Quelques chiffres...
La dystrophie musculaire Facio-Scapulo-Humérale (DMFSH) est la troisième en termes de fréquence des myopathies héréditaires, la première chez les adultes. Selon le rapport préliminaire établi par EURORDIS et Orphanet, la prévalence de la dystrophie musculaire Facio-Scapulo-Humérale est de 1/20 000. En France, on estime le nombre de patients DMFSH à environ 3250 individus. 10 à 20% d'entre eux (325-650 patients) sont en fauteuil roulant et 30 à 50% (975-1625 patients) présentent un handicap les empêchant de travailler.
Une grande variabilité d'expression intra et interfamiliale est décrite. Dans 30% des cas il n'y a pas d'antécédents familiaux connus. Les symptômes de la DMFSH apparaissent le plus souvent entre 10 et 20 ans et la maladie touche les deux sexes. Dans les cas précoces, plus rares, la maladie est plus sévère. Certains porteurs de l'anomalie génétique peuvent rester totalement asymptomatiques, ne développant aucun signe ou symptôme de la maladie.
Le tableau clinique
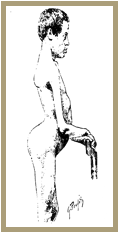
La DMFSH est caractérisée par une faiblesse des muscles de la face et de la ceinture scapulaire qui peut s'étendre progressivement aux muscles huméraux, abdominaux de la loge antérieure des jambes et à la ceinture pelvienne, souvent de manière asymétrique.
L'atteinte faciale limite l'expressivité du visage et l'occlusion complète des yeux.
L'asymétrie de l'atteinte est accentuée quand le patient sourit ("sourire transversal"). Cette atteinte, est dans 10%, des cas le seul signe de la maladie.
L'atteinte des membres supérieurs se manifeste par la saillie des omoplates ("scapula alatae") et une limitation des mouvements d'élévation des bras au-dessus de l'horizontale. L'asymétrie de l'atteinte est caractéristique et peut aider à différencier la DMFSH des autres myopathies des ceintures. Avec la progression de la maladie, en général très lente, une atteinte des muscles du bras peut être évidente.
L'atteinte des membres inférieurs touche, le plus fréquemment et à un stade précoce de la maladie, les muscles releveurs du pied ce qui entraîne des chutes et confère une démarche particulière.
La faiblesse des muscles abdominaux et de la ceinture pelvienne se manifeste simultanément dans les formes typiques avec l'atteinte des muscles huméraux.
Dans la moitié des cas, les muscles fessiers sont atteints, provoquant une bascule du bassin en avant qui, en association avec la faiblesse des muscles abdominaux, entraîne, avec une posture en hyperlordose, un abdomen proéminent.
De plus, la douleur et la fatigue sont des symptômes très fréquemment rapportés par les patients.
La génétique
La DMFSH est une maladie héréditaire. Actuellement, du point de vue génétique, il est d'usage de distinguer deux types de DMFSH : la DMFSH de type 1 (DMFSH1) et la DMFSH de type 2 (DMFSH2).
La DMFSH1 se transmet sur le mode autosomique dominant, ce qui signifie qu'un individu atteint a un risque sur deux de la transmettre à chacun de ses descendants. Cependant les cas sporadiques (absence d'apparenté atteint) sont nombreux. Ceci s'explique tout d'abord par le fait que la mutation peut survenir dans une des deux cellules sexuelles parentales à l'origine de la personne atteinte, on parle alors de mutation de novo. Il est aussi possible que la mutation survienne très tôt au cours du développement de l'embryon et soit alors présente seulement dans une partie des tissus de l'individu. On parle dans ce cas de mosaïque somatique. Cela peut aussi être lié à la pénétrance incomplète de la DMFSH, ce qui signifie que certaines personnes porteuses de l'anomalie génétique ne développent pas la maladie. Enfin, pour une même anomalie génétique héritée, l'expressivité (type et sévérité des symptômes) est variable, non seulement entre les familles, mais également entre les individus d'une même famille.
La DMFSH2 se transmet sur un mode plus complexe, que l'on peut dire "digénique", et il est important de savoir qu'à ce jour son diagnostic moléculaire n'est pas réalisé de façon systématique, il reste envisagé au cas par cas compte tenu d'un faisceau d'arguments cliniques et biologiques.
Ces deux types de DMFSH ont cependant, d'un point de vue génétique, un point commun : les deux formes de DMFSH sont liées à l'expression anormale d'un gène appelé DUX4 qui conduirait à l'expression de la maladie. Elles se différencient, en revanche, par les mécanismes qui conduisent à l'expression de ce gène DUX4.
La DMFSH1, qui concerne 95% des patients atteints, est associée à une anomalie génétique située sur le chromosome 4, dans une région appelée 4q35-qter. Dans cette région, se trouve un emplacement appelé "locus D4Z4". A ce locus, la séquence D4Z4 est normalement répétée plusieurs dizaines de fois. L'anomalie génétique présente chez les patients atteints de DMFSH1 consiste en une réduction du nombre de ces unités répétées D4Z4 (on parle de délétion ou de contraction). Cette réduction modifie l'expression de certains gènes de cette région, notamment DUX4. Le tableau clinique spécifique de la DMFSH associé à cette anomalie génétique hétérozygote, de transmission dominante et stable au fil des générations, désigne la DMFSH1.
Ainsi, chez les sujets indemnes de DMFSH1, le nombre d'unités répétées D4Z4 est supérieur ou égal à 11. Les patients atteints de DMFSH1 sont porteurs d'un nombre d'unités répétées D4Z4 compris entre 1 et 10. Il existe généralement alors une corrélation inverse entre le nombre de répétitions D4Z4 et la sévérité de la maladie : plus le nombre de répétitions est bas (en restant > 1), plus le début est précoce et les symptômes accentués. Cependant, si cette corrélation est effectivement observée à l'échelle de la population, il existe une importante variabilité entre les individus, et le nombre d'unités répétées D4Z4 chez un patient ne permet pas de prédire l'évolution chez lui ni la sévérité chez ses apparentés qui seraient porteurs de la même mutation.
Il est important de prendre en compte également d'autres facteurs génétiques qui sont associés à la DMFSH1 :
- Premièrement, la présence d'un allèle dit "permissif", est nécessaire à l'expressivité de la maladie. Il s'agit de la présence d'une séquence spécifique appelée qA dans la région subtélomérique du chromosome 4 porteur de la contraction D4Z4, en position distale. Il existe un variant de cette séquence 4qA nommé 4qB. Les deux variants, 4qA et 4qB, sont répartis avec la même fréquence dans la population générale, mais seul le variant 4qA est associé à la DMFSH. En effet, c'est l'allèle 4qA dit "permissif" qui permet l'expression stable du gène DUX4 localisé dans les unités répétées D4Z4 du chromosome 4.
Le gène DUX4 code pour une protéine facteur de transcription qui est normalement produite seulement chez l'embryon et non dans les cellules adultes différenciées. Dans ce dernier cas, l'expression du gène DUX4 est donc dite réprimée. - Deuxièmement, la contraction de la région D4Z4 conduit à un changement de la structure de l'ADN. En effet, en présence d'une réduction du nombre d'unités répétées D4Z4 comprise entre 1 et 10, nous observons un relâchement de l'ADN. Ce relâchement de l'ADN permet alors au gène DUX4 d'être accessible puis exprimé.
Pour résumer, dans le cas de la DMFSH1, la réduction du nombre d'unités répétées D4Z4 sur le chromosome 4 conduirait au relâchement de l'ADN qui permettrait alors l'expression du gène DUX4 (présent au même locus et précédemment cité).
L'expression de ce gène DUX4 conduit ensuite à la production d'un facteur de transcription qui va alors à son tour activer l'expression inopportune d'autres gènes, qui iraient alors changer le fonctionnement cellulaire normal, et conduire à la souffrance de la cellule musculaire et à l'expression de la maladie.
La DMFSH2 concerne 5 à 10% des patients qui présentent les signes cliniques de la DMFSH mais qui ne sont pas porteurs de la contraction décrite plus haut : ils présentent en effet un nombre d'unités répétées D4Z4 supérieur ou égal à 11 unités répétées.
En effet, dans le cas de la DMFSH2, d'autres mutations dans d'autres gènes ont été identifiées et notamment le gène SMCHD1 localisé sur le chromosome 18. Même si à ce jour le rôle de SMCHD1 n'est pas encore totalement connu, nous savons que dans la DMFSH2, les mutations de SMCHD1 conduisent à une hypométhylation et à un relâchement de l'ADN dans la région D4Z4 du chromosome 4. Cette modification, comme nous l'avons mentionné plus haut dans le cas de la DMFSH1, permettrait aussi l'expression aberrante du gène DUX4 (associée à un allèle permissif 4qA) qui conduirait ensuite à l'expression de la maladie DMFSH.
Pour résumer, à la différence de la DMFSH1, dans la DMFSH2, le mécanisme est indirect car il associe une mutation dans un autre gène sur un autre chromosome, notamment SMCHD1, qui induit indirectement l'expression du gène DUX4 à partir d'un allèle permissif au locus FSHD1. C'est pourquoi l'on parle alors de digénisme (deux localisations différentes sur le génome sont impliquées).
Il est important de garder en tête que, malgré la compréhension à ce jour d'une partie des origines génétiques de la DMFSH, de nombreux mécanismes modulateurs restent encore largement inconnus. De ce fait, il est parfois très difficile d'identifier, clairement et de façon fiable, des corrélations dites génotypes-phénotypes du fait de l'existence de la grande variabilité interfamiliale et interindividuelle de l'expressivité de la maladie. Cela peut alors grandement complexifier le conseil génétique en termes de prédictibilité. C'est pourquoi le diagnostic de la DMFSH doit reposer de façon prédominante sur un faisceau d'arguments prioritairement cliniques et le conseil génétique doit rester prudent.
Le diagnostic
Les diagnostics moléculaires de DMFSH sont réalisés en France dans deux laboratoires de génétique : le Service de Génétique Médicale de l'hôpital de la Timone à Marseille et le Service de Biochimie et Génétique Moléculaire de l'hôpital Cochin à Paris.
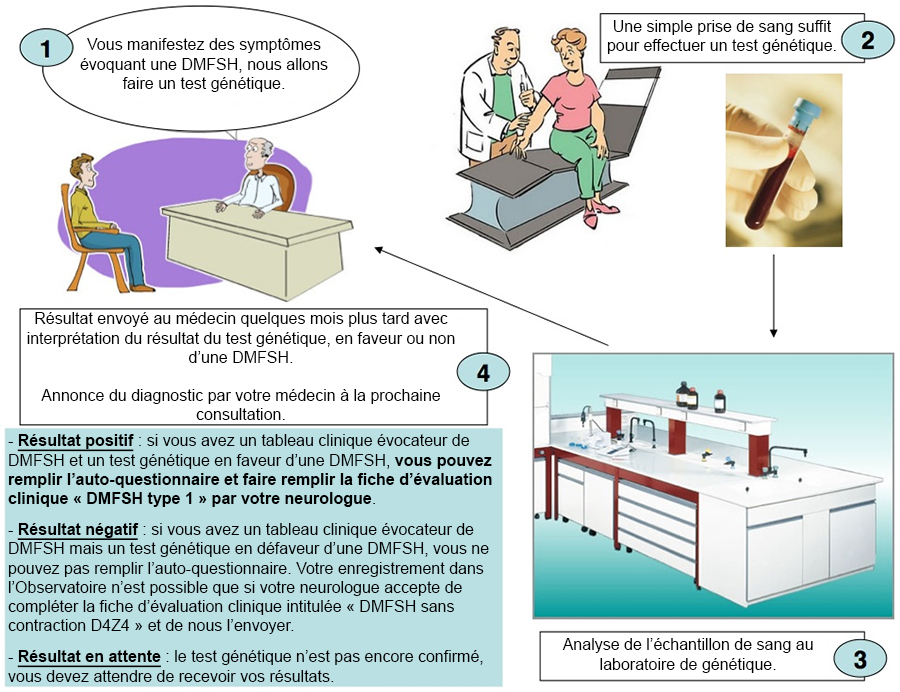
L'annonce du diagnostic dans l'étape 1 se base sur des observations cliniques. Il s'agit d'un diagnostic clinique qui doit être confirmé par un test génétique.
Le diagnostic génétique est fiable pour 95% des familles, en effet, dans un faible pourcentage de cas, environ 5%, la contraction de l'allèle D4Z4 peut être non détectée. Il parait donc nécessaire d'utiliser des techniques adaptées d'analyse diagnostique moléculaire dans la DMFSH afin d'éviter des erreurs diagnostiques.
Éducation du patient
PROSOL E-learning
PROSOL E-learning est une plateforme de formation, libre et gratuite, qui traite des maladies neurologiques entrainant des troubles moteurs, sensitifs et cognitifs. Le site internet propose trois parcours de formation, traduits en anglais et en italien. Cette plateforme est en constante évolution, et a vocation à s'étendre à d'autres thématiques médicales.
Programme d'éducation thérapeutique du patient (ETP)
Le programme d'éducation thérapeutique organisé par le Centre de référence des maladies rares neuromusculaires (CRMR NM) du CHU de Nice s'adresse aux patients atteints de dystrophie musculaire facio-scapulo-humérale (FSH). Il comprend des ateliers qui visent à permettre aux participants d'acquérir de nouvelles connaissances et compétences sur leur pathologie pour améliorer leur quotidien et leur qualité de vie. Ces ateliers collectifs sont animés par l'équipe pluridisciplinaire du Centre de référence qui se compose de neurologue, kinésithérapeute, ergothérapeute, diététicienne, psychologue. Le programme est proposé lors d'une consultation simple ou pluridisciplinaire. Le médecin et le patient établissent ensemble le bilan éducatif partagé. En fonction de ses besoins, le patient peut s'inscrire aux ateliers souhaités qui lui permettront d'échanger avec les praticiens et d'autres patients, atteints de la même maladie.
Les domaines abordés sont :
- Connaître la pathologie, les nouvelles stratégies thérapeutiques et les ressources à disposition
- Choisir une activité physique adaptée
- Gérer les difficultés dans les activités de la vie quotidienne
- Gérer son état nutritionnel
- Réguler ses activités pour gérer la fatigue et son impact psychologique
Pour plus d'informations sur le programme, contactez la psychologue du centre :
Sophie Meiran
E-mail : meiran.s@chu-nice.fr
Téléphone : 04 92 03 72 42